CASE REVIEW
Early screening for familial hypercholesterolemia helps optimize the additional LDL-lowering benefit of add-on PCSK9 inhibitor



Familial hypercholesterolemia (FH) is a common codominant monogenic dyslipidemia that can result in the lifelong and prominent elevation of plasma low-density lipoprotein cholesterol (LDL-C).1,2 Whereas homozygous FH is rare with an estimated frequency of 1 in 160,000-320,000 individuals, heterozygous FH is more prevalent with 1 in 200-250 persons being affected.1 Yet, FH remains largely undetected and hence undertreated in Hong Kong, leading to a heightened risk of premature atherosclerotic cardiovascular disease (ASCVD) such as coronary heart disease (CHD).2 In a recent interview with Omnihealth Practice, Professor Tan, Choon-Beng Kathryn, Sir David Todd Professor in Medicine, Department of Medicine, The University of Hong Kong, called for clinicians’ earlier actions to screen families with index FH cases for more timely initiation of cholesterol-lowering therapies. She also advised clinicians to consider a more aggressive LDL-C-reduction strategy including the addition of the newer proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitor, if indicated, through the sharing of 2 local cases here.
Background
In Hong Kong, FH remains a largely underdiagnosed and undertreated condition due to the infeasibility of universal FH screening and the difficulty of convincing asymptomatic patients to participate in general screening.2 While no well-established population data is available in Hong Kong, it has been estimated that only 2-3% of FH cases are actually diagnosed, with no foreseeable improvement in near future.3
To increase the likelihood of identifying the very first index FH cases, a plasma cholesterol profile with a well-documented family history of hypercholesterolemia and/or premature CHD should be integrated into routine testing.2 When evaluating the cholesterol profile, adults having a plasma LDL-C level >5mmol/L should be regarded as possible probands after excluding secondary causes of hypercholesterolemia.2 For high-risk patients who have a family history of FH or premature CHD, an LDL-C level of 4.5mmol/L should be the threshold for FH suspicion.2 Checking for the presence of tendon xanthomas is also important as it is a specific sign for FH.2
Once there is a high probability suggesting a diagnosis of FH, diagnostic scoring systems are available to help to make a clinical diagnosis based on phenotype.4 Typically, the Dutch Lipid Clinic Network (DLCN) Criteria and the Simon Broome Criteria are used in clinical practice for diagnosing FH.4 In Hong Kong, concerted expert opinion and consensus recommended the use of the DLCN Criteria (Figure 1).2,4 Genetic testing is not widely available in the public sector, and genetic testing is mainly used to confirm diagnosis where there is a strong likelihood of FH based on clinical criteria.
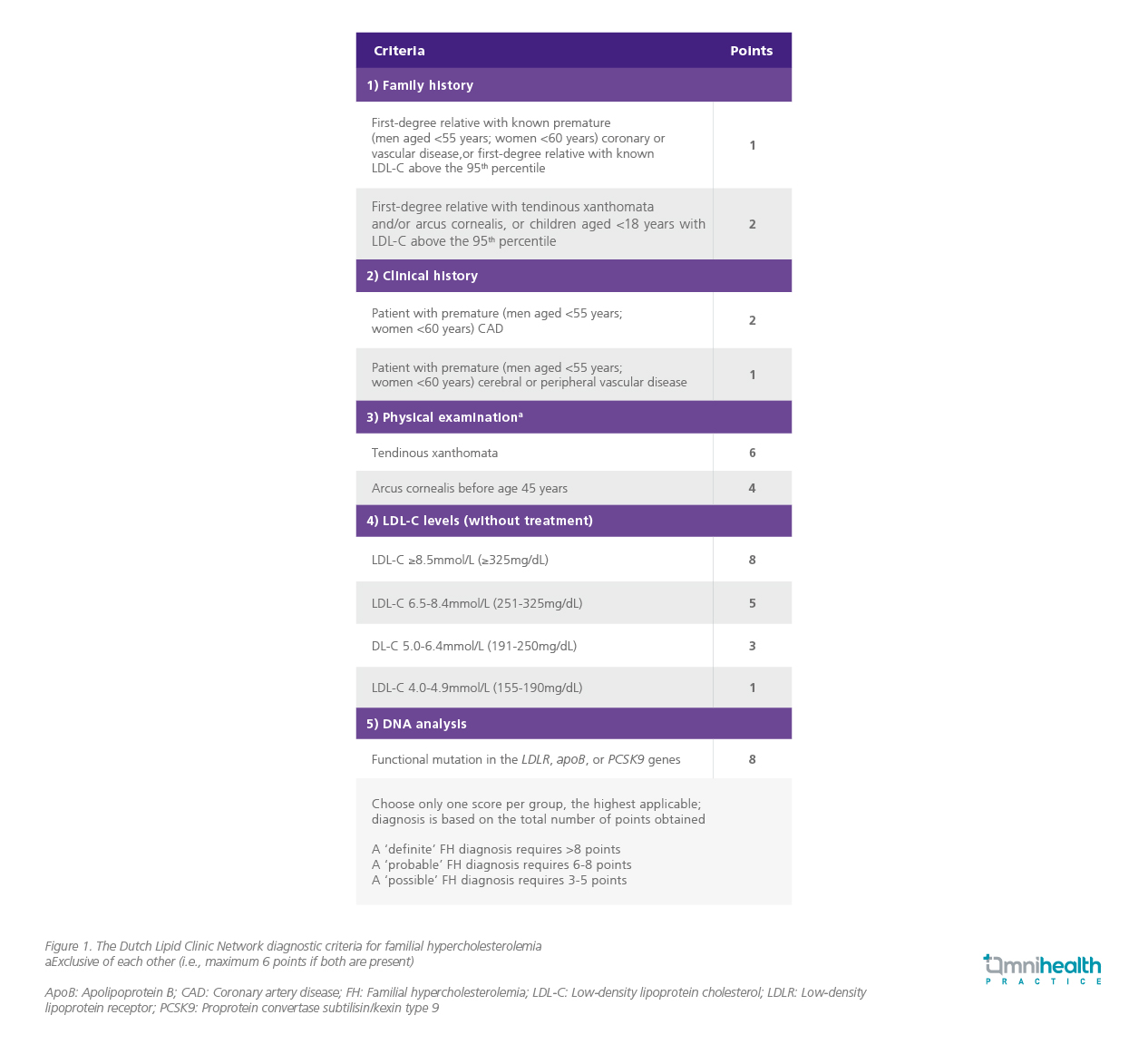
Upon identifying index cases, clinicians should proceed with cascade screening for the rest of the family members as this is the most cost-effective approach in discovering additional FH cases.2,4 Similar to diagnosing index cases, cascade family screening can encompass plasma lipid profiles and genetic testing.2 From clinical experience, most asymptomatic family members are reluctant to undergo screening. Therefore, clinicians have a responsibility to advise family members of their elevated FH risk and the need of proactive screening to prevent potential cardiovascular disease.5
As the prognosis of FH is highly dependent on the plasma LDL-C level, maintaining a sufficiently low LDL-C level should be the primary treatment objective for FH patients.2 As such, cholesterol-lowering treatments should be started instantaneously after a FH diagnosis has been established.1 Specifically for the FH patients in Hong Kong, while a general LDL-C target of <2.5mmol/L is suggested for primary CHD prevention, a more stringent level of <1.8mmol/L is required for patients who are at very-high risk of ASCVD.2 The latest guidelines from the European Society of Cardiology recommend that patients with a very high risk of ASCVD are subjected to a much stricter LDL-C target of <1.4mmol/L and ≥50% reduction from baseline.1
To monitor ASCVD risk for better treatment optimization, imaging techniques such as computed tomographic coronary angiography and carotid ultrasound imaging that can detect asymptomatic atherosclerosis can be employed.1,2 From clinical experience, imaging techniques can help to identify the presence of underlying cardiovascular diseases and the need for more aggressive treatment strategies. Typically, patients are initiated with high-intensity statins followed by gradual dose titration when necessary.2 If the target LDL-C level cannot be reached with statin monotherapy, adding ezetimibe and/or bile-acid sequestrant or niacin to the regimen is recommended.2 However, in light of the very stringent LDL-C goal established by guidelines, a <1.4mmol/L target may not be easily achieved even with maximal tolerated statin plus ezetimibe and would require more potent therapies such as the PCSK9 inhibitor.1 In addition, patients who are considered statin intolerant are unlikely to achieve the guideline-recommended LDL-C level with ezetimibe alone, which highlights the need for PCSK9 inhibitor when aggressive therapy is required.1,6
Case report
To illustrate the use of PCSK9 inhibitor in real-world practice, Prof. Tan shared two of her cases.
Case 1:
Heterozygous FH Person A is a 40-year-old male who was diagnosed to have FH at the age of 17 years during family screening. The index case was his elder brother who presented with premature cardiovascular disease in his 20s. Initial diagnosis revealed the patient had a very high LDL-C level of 13.4mmol/L. He had tendon xanthomas but no clinical evidence of cardiovascular disease. Subsequent genetic testing confirmed the diagnosis of heterozygous FH with mutation in the LDL receptor gene.
He was initiated with atorvastatin at the time of diagnosis and later up titrated to a maximum dose with ezetimibe and cholestyramine as addon therapy. Despite the combined use of three lipid-lowering agents, his LDL-C level remained above target at 5.1mmol/L. He refused apheresis and was started on PCSK9 inhibitor in 2016 when the drug became available in Hong Kong. Overall, the 4-drug regimen had lowered his LDL-C level from a baseline of 13.4mmol/L to 3.0mmol/L. Until now, he remains asymptomatic and experiences no major intolerable adverse effects.
Case 2:
Homozygous FH Born to parents with known FH, person B was diagnosed with homozygous FH early at 12-years-old. Initial diagnosis indicated an LDL-C level of 9.3mmol/L and tendon xanthomas. He received treatment at pediatric department during his childhood but defaulted all subsequent follow-up visits after turning 18-years-old. He presented with a myocardial infarction (MI) at the age of 35 and required percutaneous coronary intervention (PCI). After his MI was treated, he was given high-dose rosuvastatin and ezetimibe, which lowered his LDL-C to 5.4mmol/L. While his LDL-C remained off-target, he also declined apheresis and he remained on the 2-drug regimen until the advent of PCSK9 inhibitor. After incorporating a PCSK9 inhibitor, his LDL-C was further reduced to 3.2mmol/L. Overall, he responded well to the PCSK9 inhibitor with no notable major adverse events.
Discussion
From the shared cases, not only could PCSK9 inhibitors substantially reduce the LDL-C level on top of other cholesterol-lowering therapies, the LDL-C lowering effect could also be sustained for a long period of time. “Indeed, the patients were pleased to see their LDL-C levels reduced to an unprecedented 3mmol/L after the initiation of PCSK9 inhibitor,” Prof. Tan commented. The efficacy of PCSK9 inhibitors in reducing LDL-C levels is comparable among heterozygous FH patients with a variety of underlying genetic mutations, whereas the effectiveness of PCSK9 inhibitor in homozygous FH is partly determined by the underlying genetic abnormality. When both alleles of the LDL receptor gene are affected by a null mutation, very little response is observed.7
Despite the impressive efficacy of PCSK9 inhibitor as shown in clinical trials and real-world data, the merit of significant LDL-C reduction still hinges on the identification of the disease in the first place. Therefore, it is clinically important to make an early diagnosis of FH, which can be achieved through the opportunistic screening of high-risk individuals. Once an index case is confirmed, his/her family should be screened without procrastination. As seen in case 1, owing to the elder brother’s medical history that led to the family screening of person A, the patient benefited from early treatments that had successfully kept him asymptomatic of any cardiovascular diseases from the age of FH diagnosis at 17 till 40-years-old.
Once a diagnosis of FH is made, LDL-C lowering treatments should be initiated immediately in addition to lifestyle measures, focusing on an individualized titration regimen and subsequent add-on therapy according to the target LDL-C level.8 Importantly, a delay of timely and appropriate treatments can put patients at risk of CHD. As evidenced in case 2, despite person B’s early FH diagnosis at 12-years-old, his non-adherence to follow-up visits and treatments resulted in a MI at an unusually young age. “Had person B not defaulted follow-up visits and adhered to his treatments, he probably wouldn’t have experienced a MI at 35-years-old,” Prof. Tan reflected. Thus, early treatment initiation with statin as well as subsequent regimen titration and the prompt use of add-on therapy, such as the PCSK9 inhibitor, is pivotal to achieving the LDL-C target, to not only manage any established diseases but also reduce the future risks of cardiovascular events.
Conclusion
Due to the lack of universal screening and the reluctance of families to undergo screening, FH remains a highly undetected condition in clinical practice. Clinicians should thus take the initiative to perform opportunistic screening for individuals at high risk for FH based on LDL-C level and family history of premature CHD and/or hypercholesterolemia. After the identification of an index case, cascade screening of the family members should be carried out to identify new cases of FH. Once a diagnosis is made, treatments should be initiated immediately. Further dosage titration and add-on therapy may also be necessary, depending on the LDL-C goal and patients’ ASCVD risk. Since PCSK9 inhibitor can significantly reduce LDL-C up to 60% on top of other cholesterol-lowering therapies, this has offered patients a better chance of treatment goal attainment and CHD prevention.
This is an independent editorial article, published and distributed through unrestricted educational support from Amgen Hong Kong Limited, for the purpose of continuing medical education only. The views expressed in this publication reflect the experience and/or opinion of the author(s) and are not necessarily those of editors, publisher and sponsor(s). Because of rapid advances in medicine, independent verification of clinical diagnoses, medical suitability and dosage should be made before treatment prescription. The appearance of advertisement, if any, has no influence on editorial content or presentation and does not imply the endorsement of products by the publication, or its authors and editors.
- Mach F et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J. 2020;41:111-188.
- Tomlinson B et al. Guidance on the management of familial hypercholesterolaemia in Hong Kong: an expert panel consensus viewpoint. Hong Kong Med J. 2018;24(4):408-415.3.
- Pang J et al. Comparative aspects of the care of familial hypercholesterolemia in the “Ten Countries Study”. J Clin Lipidol. 2019;13(2):287-300.
- Tan K et al. Genetic screening for familial hypercholesterolaemia in Hong Kong. Hong Kong Med J. 2018;24(3):7-10.5.
- Chan ML et al. Genetic variations in familial hypercholesterolemia and cascade screening in East Asians. Mol Genet Genomic Med. 2019;7(2):e00520.
- Koba S et al. Evolocumab vs. ezetimibe in statin-intolerant hyperlipidemic Japanese patients: phase 3 GAUSS-4 trial. J Atheroscler Thromb. 2020;27(5):471-484.
- Brandts J et al. A meta-analysis of medications directed against PCSK9 in familial hypercholesterolemia. Atherosclerosis. 2021;325:46-56.
- Lui DT et al. Management of familial hypercholesterolemia: current status and future perspectives. J Endo Soc. 2021;5:1-14.

