FEATURES
Shaping the future of myelofibrosis care: The role of momelotinib in addressing anemia, spleen and symptom burden in once-daily oral regimen



Myelofibrosis (MF) is a hematologic disorder characterized by splenomegaly, constitutional symptoms, ineffective hematopoiesis, and an increased risk of leukemic transformation.1,2 While Janus kinase (JAK) inhibitors have transformed MF care, most are associated with anemia and increased transfusion dependence, which impacts survival—leaving a critical treatment gap.2 At the recent symposium on “The Management of Myelofibrosis with Anaemia”, hosted by The Hong Kong Society of Haematology, Professor Haifa Kathrin Al-Ali, from the University Hospital of Halle (Saale), Germany, joined Dr. Gill Harinder Singh Harry, from the University of Hong Kong, to discuss emerging therapeutic strategies in MF.
Myelofibrosis – A heterogeneous disease requiring a personalized approach
MF is a clonal hematopoietic stem cell disorder marked by constitutional symptoms, extramedullary hematopoiesis leading to splenomegaly, and a risk of progression to blast-phase disease or acute myeloid leukemia (AML).1-3 Given its heterogeneity, a personalized treatment approach is crucial, as patients experience varying degrees of constitutional symptoms, splenomegaly, and cytopenias.1,2,4
While thrombocytopenia is relatively uncommon at diagnosis, 10%-15% of patients develop it over time.2,4,5 In contrast, anemia is more prevalent, affecting more than one-third of patients at diagnosis and worsening as the disease progresses.2,5 MF-related anemia is primarily driven by ineffective erythropoiesis, often exacerbated by cytoreductive therapy and JAK inhibitors such as ruxolitinib, with additional contributions from less common causes such as bleeding, immune-mediated hemolysis, or hematinic deficiencies.2,6 Anemia in MF is more than just a symptom—it is a key prognostic factor that affects both survival and quality of life.3,6 Given its impact, emerging therapies are being explored to address this critical gap.2,4,5
The expanding JAK inhibitor landscape in MF
JAK inhibitors remain the cornerstone of MF management, offering disease control and symptom relief.2-4 Ruxolitinib, a non-selective JAK1/2 inhibitor, reduces cellular proliferation and splenomegaly by targeting constitutive JAK/STAT activation while alleviating constitutional symptoms through cytokine suppression.1,4 “Ruxolitinib is well established for controlling splenomegaly and cytokine-related symptoms, with pooled analyses suggesting a survival benefit,” noted Dr. Gill.
The COMFORT-I and COMFORT-II trials demonstrated that ruxolitinib significantly prolonged overall survival (OS), with earlier initiation (≤12 months from diagnosis) associated with improved spleen response, longer OS, and fewer hematologic toxicities.1,4 However, its non-selective JAK1/2 inhibition often leads to dose-limiting anemia and thrombocytopenia, impacting long-term efficacy.2,4 Furthermore, approximately 50% of patients discontinue ruxolitinib by year three, often due to disease progression, suboptimal response, or cytopenias.1,4
To better predict outcomes, the Response to Ruxolitinib After 6 Months (RR6) model was developed to prognosticate survival for patients with MF after 6 months of ruxolitinib treatment.7 This model incorporates several parameters: ruxolitinib dose intensity (<20mg twice daily at baseline, 3 and 6 months) [1 point], red blood cell (RBC) transfusion requirement at 3 and/or 6 months [1 point], spleen response (≤30% reduction in palpable length) with respect to baseline at months 3 and 6 [1.5 points], and RBC transfusion requirement at all time points [1.5 points] (table 1).7
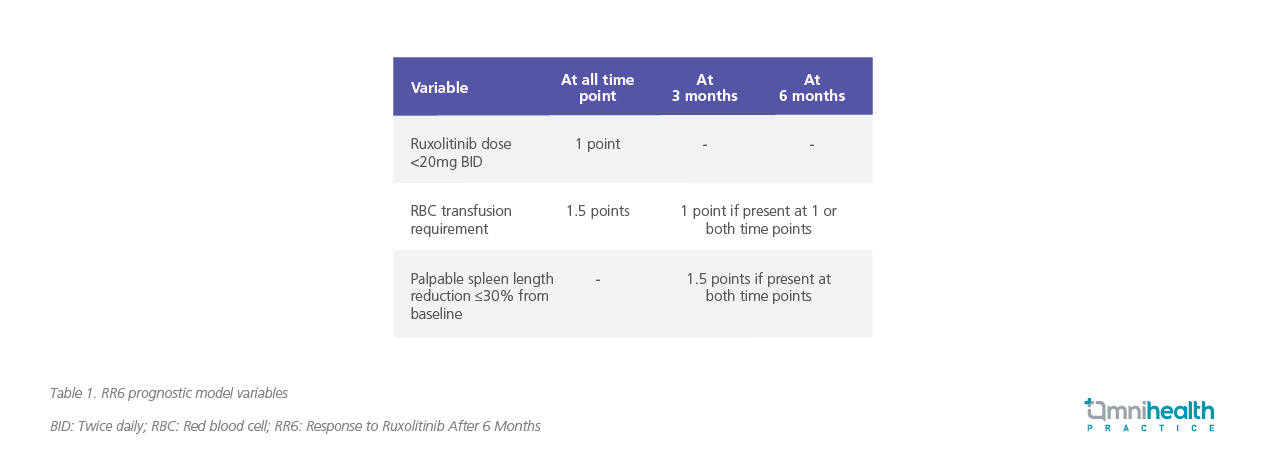
Patients are classified into three risk categories with distinct prognoses: low-risk (score 0; median OS not reached); intermediate-risk (score 1-2; median OS of 61 months); and high-risk (score ≥2.5; median OS of 33 months) (table 2).7
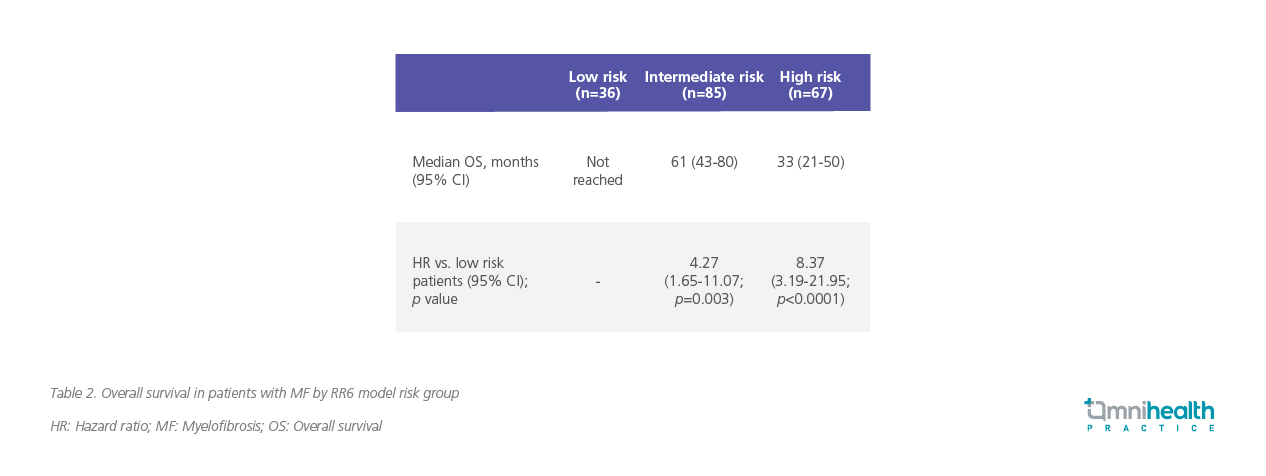
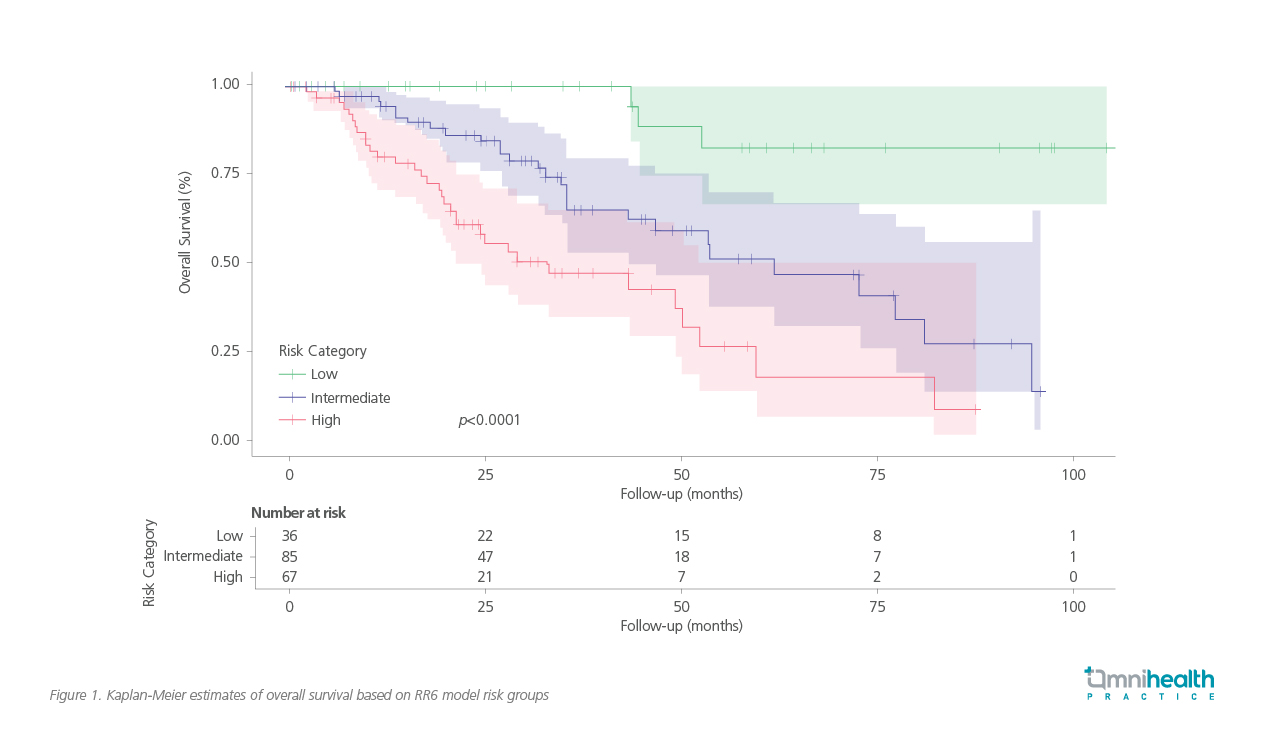
A Kaplan-Meier analysis further illustrates the survival differences across RR6 risk groups, reinforcing the model’s ability to identify patients with suboptimal response early in their treatment course (figure 1).7 “Poor outcomes are expected once ruxolitinib is discontinued, particularly in patients with high-risk genetic profiles,” he added, highlighting the need to proactively evaluate treatment response before disease progression necessitates discontinuation.
To address the unmet needs in MF management, next-generation JAK inhibitors have been developed with distinct mechanisms to overcome limitations from earlier therapies.1 Momelotinib stands out for its dual inhibition of JAK1/2 and ACVR1, a key regulator of SMAD2/3 signaling involved in hepcidin production and iron-restricted erythropoiesis, both major drivers of MF-related anemia.1,2,4 By targeting ACVR1, momelotinib reduces hepcidin levels, helping to improve anemia, while enabling effective JAK inhibition.2,4,6 With its ability to address anemia, symptom burden and splenomegaly, momelotinib represents a promising option for both JAK-naïve and JAK-exposed patients who struggle with anemia and transfusion dependence.2
Momelotinib in JAK inhibitor-naïve and experienced in MF patients: A focus on anemia
Momelotinib has been evaluated in phase 3 trials, both in first-line and second-line settings.8,9 The phase 3 SIMPLIFY-1 trial (n=432) was a randomized, double-blind, noninferiority study comparing the efficacy and safety of momelotinib to ruxolitinib in JAK inhibitor-naïve patients with MF.8 A post-hoc efficacy analysis focused on patients with baseline Hb <10g/dL (n=181) demonstrated momelotinib’s noninferiority to ruxolitinib in the proportion of patients achieving ≥35% spleen volume reduction (SVR35) at week 24 (31% vs. 33%).10
While momelotinib showed a relatively lower rate of total symptom score reduction of ≥50% (TSS50) compared to ruxolitinib (25% vs. 36%), it demonstrated a clear advantage in improving anemia, with a higher proportion of patients achieving transfusion independence (TI) (67% vs. 49%).6,8,10 This is particularly meaningful, as anemia remains a major contributor to reduced survival and quality of life in patients with MF, even when SVR is limited.3,4,6 “Even in cases where spleen reduction is modest, the ability to improve anemia translates into meaningful clinical benefits,” noted Dr. Gill. These insights sparked further exploration in the MOMENTUM trial, which evaluated momelotinib’s efficacy in JAK inhibitor-experienced patients with symptomatic anemia.9
MOMENTUM’s defining impact on TI and improved OS
MOMENTUM was the first phase 3 randomized, double-blind trial in MF to use symptom improvement as its primary endpoint, marking a shift in clinical trial design.9 The trial evaluated momelotinib vs. danazol in JAK inhibitor-experienced MF patients with symptomatic anemia (n=195).9 Participants had moderate-to-severe anemia (Hb <10g/dL) and splenomegaly, with ~80% requiring RBC transfusion within 8 weeks prior to enrolment in both treatment groups.9 The trial’s primary endpoint was the Myelofibrosis Symptom Assessment Form (MFSAF) TSS50 response rate at week 24, while key secondary endpoints included TI rate, splenic response rates, and rate of zero transfusion at week 24.9 TSS50 was achieved in 25% of momelotinib-treated patients vs. 9% with danazol (p=0.0095).9 Momelotinib also improved TI rate from 13% at baseline to 31% at week 24 vs. from 15% to 20% with danazol (p=0.0064) and achieved significant SVR35 (23% vs. 3%; p=0.0006), with durable responses through week 48.9,11
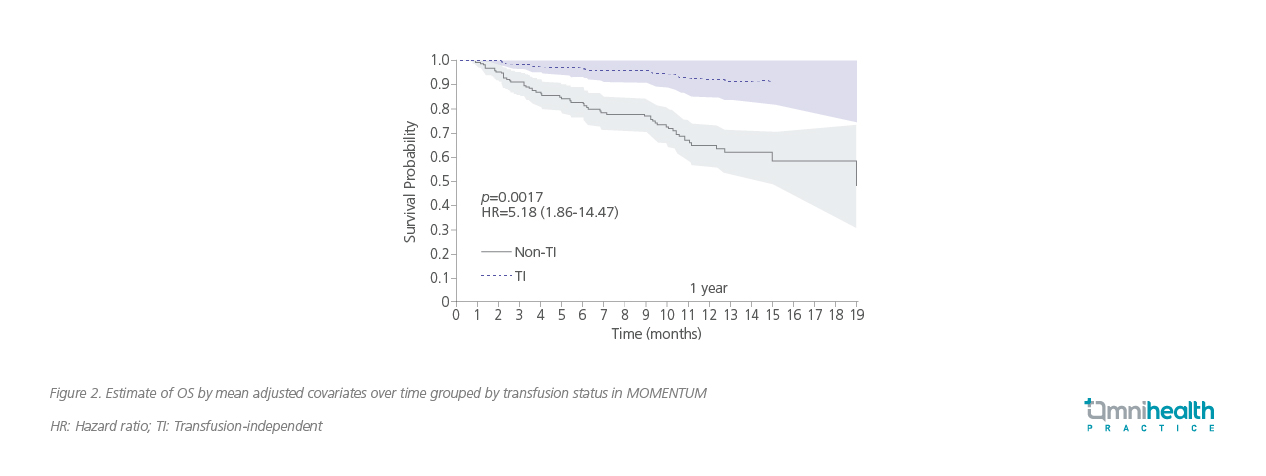
The discussion then shifted toward the importance of achieving TI, an outcome that significantly impacts both quality of life and long-term prognosis.3,6 “The MOMENTUM trial was designed specifically to address this,” Prof. Al-Ali noted. “It showed that momelotinib not only reduces the need for transfusions but does so without compromising symptom control or spleen response”. A post-hoc time-dependent analysis also revealed that TI strongly correlated with improved OS (figure 2).12
In SIMPLIFY-1 and MOMENTUM, patients were able to transition to momelotinib during the open-label extension phases without a washout period or treatment overlap.13 Dr. Gill noted that while no formal guidelines exist for transitioning therapies, this approach was feasible in the clinical trial setting. Notably, momelotinib does not require dose adjustment in patients with severe thrombocytopenia prior to treatment initiation.2
This contrasts with ruxolitinib, which requires dose modifications in patients with anemia or thrombocytopenia.4,8 By comparison, momelotinib offers the convenience of a simple, once-daily dose at full strength—even in patients with baseline thrombocytopenia.2,10 This streamlined dosing regimen ensures consistent efficacy without the need for dose adjustments.2 As MF treatment continues to evolve, real-world insights reinforce momelotinib’s role in addressing anemia, improving TI rates, and offering a simplified, tolerable regimen for patients with MF.14
Conclusion
Momelotinib is emerging as a pivotal treatment option for MF, particularly in patients with moderate-to-severe anemia as highlighted by the SIMPLIFY-1 trial.10 The MOMENTUM trial further demonstrated significant improvements in TI, addressing a key unmet need in MF management.2,9 Beyond merely spleen reduction, momelotinib’s unique mechanism offers broader disease control, reinforcing the shifting treatment paradigm—one that prioritizes functional independence, symptom relief, and overall quality of life.2,6,9 With its simplified, full-dose once-daily regimen and seamless treatment transition, momelotinib provides a practical solution in clinical settings.9,10,13,14

